animalcules
Yue Zhao
Boston University School of Medicine, Boston, MABioinformatics Program, Boston University, Boston, MAAnthony Federico
Boston University School of Medicine, Boston, MABioinformatics Program, Boston University, Boston, MAEvan Johnson
Boston University School of Medicine, Boston, MABioinformatics Program, Boston University, Boston, MAJanuary 22, 2024
Source:vignettes/animalcules.Rmd
animalcules.RmdCitation
Note: if you use animalcules in published research, please cite:
Zhao, Y., Federico, A., Faits, T. et al. (2021) animalcules: interactive microbiome analytics and visualization in R Microbiome, 9(1), 1-16. 10.1186/s40168-021-01013-0
Introduction
animalcules is an R package for utilizing up-to-date data analytics, visualization methods, and machine learning models to provide users an easy-to-use interactive microbiome analysis framework. It can be used as a standalone software package or users can explore their data with the accompanying interactive R Shiny application. Traditional microbiome analysis such as alpha/beta diversity and differential abundance analysis are enhanced, while new methods like biomarker identification are introduced by animalcules. Powerful interactive and dynamic figures generated by animalcules enable users to understand their data better and discover new insights.
Installation
Install the development version of the package from Bioconductor.
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("wejlab/animalcules")Or install the development version of the package from Github.
if (!requireNamespace("devtools", quietly = TRUE)) {
install.packages("devtools")
}
devtools::install_github("wejlab/animalcules")Load Packages
Load the packages needed into R session.
## Registered S3 method overwritten by 'httr':
## method from
## print.response rmutil## Loading required package: MatrixGenerics## Loading required package: matrixStats##
## Attaching package: 'MatrixGenerics'## The following objects are masked from 'package:matrixStats':
##
## colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
## colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
## colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
## colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
## colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
## colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
## colWeightedMeans, colWeightedMedians, colWeightedSds,
## colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
## rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
## rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
## rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
## rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
## rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
## rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
## rowWeightedSds, rowWeightedVars## Loading required package: GenomicRanges## Loading required package: stats4## Loading required package: BiocGenerics##
## Attaching package: 'BiocGenerics'## The following objects are masked from 'package:stats':
##
## IQR, mad, sd, var, xtabs## The following objects are masked from 'package:base':
##
## anyDuplicated, aperm, append, as.data.frame, basename, cbind,
## colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
## get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply,
## match, mget, order, paste, pmax, pmax.int, pmin, pmin.int,
## Position, rank, rbind, Reduce, rownames, sapply, setdiff, sort,
## table, tapply, union, unique, unsplit, which.max, which.min## Loading required package: S4Vectors##
## Attaching package: 'S4Vectors'## The following object is masked from 'package:utils':
##
## findMatches## The following objects are masked from 'package:base':
##
## expand.grid, I, unname## Loading required package: IRanges## Loading required package: GenomeInfoDb## Loading required package: Biobase## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.##
## Attaching package: 'Biobase'## The following object is masked from 'package:MatrixGenerics':
##
## rowMedians## The following objects are masked from 'package:matrixStats':
##
## anyMissing, rowMediansRun Shiny App
This command is to start the animalcules shiny app. For shiny app tutorials, please go to our website, and click the tab “Interactive Shiny Analysis”.
The rest of the vignette is for the command line version of animalcules, which could also be found in our website, and click the tab “Command Line Analysis”.
Load Toy Dataset
This toy dataset contains 50 simulated samples. Throughout the vignette, we will use the data structure MultiAssayExperiment (MAE) as the input to all functions.
data_dir <- system.file("extdata/TB_example_dataset.rds",
package = "animalcules")
MAE <- readRDS(data_dir)For users who would like to use their own dataset, please follow the following steps:
- Use the shiny app to upload your dataset. [check shiny tutorial] (https://wejlab.github.io/animalcules-docs/articles/rmd/upload.html)
- Go to tab 2 “Data Summary and Filtering”, and click the button “Download Animalcules File”. [check shiny tutorial] (https://wejlab.github.io/animalcules-docs/articles/rmd/summary.html)
- Load the downloaded .rds animalcules file and rename it as MAE in R.
data_dir <- "PATH_TO_THE_ANIMALCULES_FILE"
MAE <- readRDS(data_dir)Summary and Categorize
One of the first steps in the data analysis process involves summarizing the data and looking for outliers or other obvious data issues that may cause issue with downstream analysis.
Summary Plot (Pie Chart or Box plot)
One type of summarization plot returns either a box plot or pie chart for continous or categorical data respectively.
p <- animalcules::filter_summary_pie_box(MAE,
samples_discard = c("SRR1204622"),
filter_type = "By Metadata",
sample_condition = "age_s"
)
pSummary Plot (Density Plot or Bar Plot)
One type of summarization plot returns either a density plot or bar plot for continous or categorical data respectively.
p <- filter_summary_bar_density(MAE,
samples_discard = c("SRR1204622"),
filter_type = "By Metadata",
sample_condition = "sex_s"
)
pCategorize
It is often necessary to bin continuous data into categories when performing analyses that require categorical input. To help ease this process, users can automatically categorize categorical data and provide custom bin breaks and labels in doing so.
microbe <- MAE[["MicrobeGenetics"]]
samples <- as.data.frame(colData(microbe))
result <- filter_categorize(samples,
sample_condition = "age_s",
new_label = "AGE_GROUP",
bin_breaks = c(0, 30, 40, 100),
bin_labels = c("a", "b", "c")
)
head(result$sam_table)## isolate_s Disease age_s sex_s tissue_s AGE_GROUP
## SRR1204622 Patient 1 TB 45 F Sputum c
## SRR1204623 Patient 1 TB 45 F Nasal c
## SRR1204624 Patient 1 TB 45 F Oropharynx c
## SRR1204625 Patient 2 TB 29 M Sputum a
## SRR1204626 Patient 2 TB 29 M Nasal a
## SRR1204627 Patient 2 TB 29 M Oropharynx a
result$plot.unbinned
result$plot.binnedVisualization
A typical analysis involves visualization of microbe abundances across samples or groups of samples. Animalcules implements three common types of visualization plots including stacked bar plots, heat map, and box plots generated with Plotly.
Relative Abundance Stacked Bar Plot
The stacked bar plots are used to visualize the relative abundance of microbes at a given taxonomical level in each sample represented as a single bar.
p <- relabu_barplot(MAE,
tax_level = "genus",
sort_by = "conditions",
sample_conditions = c("Disease"),
show_legend = TRUE
)
pRelative Abundance Heatmap
The heatmap represents a sample by organisms matrix that can be visualized at different taxonomic levels.
p <- relabu_heatmap(MAE,
tax_level = "genus",
sort_by = "conditions",
sample_conditions = c("sex_s", "age_s")
)
pRelative Abundance Boxplot
The boxplot visualization allows users to compare the abundance of one or more organisms between categorical attributes.
p <- relabu_boxplot(MAE,
tax_level = "genus",
organisms = c("Streptococcus", "Staphylococcus"),
condition = "sex_s",
datatype = "logcpm"
)## Using Row.names, sex_s as id variables
p## Warning in RColorBrewer::brewer.pal(N, "Set2"): minimal value for n is 3, returning requested palette with 3 different levels
## Warning in RColorBrewer::brewer.pal(N, "Set2"): minimal value for n is 3, returning requested palette with 3 different levels## Warning: 'layout' objects don't have these attributes: 'boxmode'
## Valid attributes include:
## '_deprecated', 'activeshape', 'annotations', 'autosize', 'autotypenumbers', 'calendar', 'clickmode', 'coloraxis', 'colorscale', 'colorway', 'computed', 'datarevision', 'dragmode', 'editrevision', 'editType', 'font', 'geo', 'grid', 'height', 'hidesources', 'hoverdistance', 'hoverlabel', 'hovermode', 'images', 'legend', 'mapbox', 'margin', 'meta', 'metasrc', 'modebar', 'newshape', 'paper_bgcolor', 'plot_bgcolor', 'polar', 'scene', 'selectdirection', 'selectionrevision', 'separators', 'shapes', 'showlegend', 'sliders', 'smith', 'spikedistance', 'template', 'ternary', 'title', 'transition', 'uirevision', 'uniformtext', 'updatemenus', 'width', 'xaxis', 'yaxis', 'barmode', 'bargap', 'mapType'Diversity
Alpha Diversity Boxplot
Alpha diversity, which describes the richness and evenness of sample microbial community, is a vital indicator in the microbiome analysis. animalcules provides the interactive boxplot comparison of alpha diversity between selected groups of samples. Both taxonomy levels and alpha diversity metrics (Shannon, Gini Simpson, Inverse Simpson) can be changed. Users can also conduct alpha diversity statistical tests including Wilcoxon rank sum test, T test and Kruskal-Wallis test.
Plot the alpha diversity boxplot between the levels in selected condition.
alpha_div_boxplot(
MAE = MAE,
tax_level = "genus",
condition = "Disease",
alpha_metric = "shannon"
)Alpha Diversity Statistical Test
Conduct statistical test on the alpha diversity between the levels in selected condition.
do_alpha_div_test(
MAE = MAE,
tax_level = "genus",
condition = "Disease",
alpha_metric = "shannon",
alpha_stat = "T-test"
)## Wilcoxon rank sum exact test Welch Two Sample t-test
## P-value 0.006682257 0.006483754Beta Diversity Heatmap
On the other hand, by defining distances between each sample, beta diversity is another key metric to look at. Users can plot the beta diversity heatmap by selecting different beta diversity dissimilarity metrics including Bray-Curtis and Jaccard. Users can also conduct beta diversity statistical testing between groups including PERMANOVA, Wilcoxon rank sum test and Kruskal-Wallis.
Plot the beta diversity heatmap with selected condition.
diversity_beta_heatmap(
MAE = MAE,
tax_level = "genus",
input_beta_method = "bray",
input_bdhm_select_conditions = "Disease",
input_bdhm_sort_by = "condition"
)## No scatter mode specifed:
## Setting the mode to markers
## Read more about this attribute -> https://plotly.com/r/reference/#scatter-modeBeta Diversity Boxplot
Plot the beta diversity boxplot within and between conditions.
diversity_beta_boxplot(
MAE = MAE,
tax_level = "genus",
input_beta_method = "bray",
input_select_beta_condition = "Disease"
)Beta Diversity Test
Conduct statistical test on the beta diversity between the levels in selected condition.
diversity_beta_test(
MAE = MAE,
tax_level = "genus",
input_beta_method = "bray",
input_select_beta_condition = "Disease",
input_select_beta_stat_method = "PERMANOVA",
input_num_permutation_permanova = 999
)## Permutation test for adonis under reduced model
## Terms added sequentially (first to last)
## Permutation: free
## Number of permutations: 999
##
## vegan::adonis2(formula = dist.mat ~ condition, data = sam_table, permutations = input_num_permutation_permanova)
## Df SumOfSqs R2 F Pr(>F)
## condition 1 1.1942 0.13939 4.535 0.003 **
## Residual 28 7.3730 0.86061
## Total 29 8.5671 1.00000
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Beta Diversity NMDS Plot
Display an NMDS plot based on the selected beta diversity analysis performed.
diversity_beta_NMDS(
MAE = MAE,
tax_level = "genus",
input_beta_method = "bray",
input_select_beta_condition = "Disease"
)## Run 0 stress 0.1231453
## Run 1 stress 0.1495791
## Run 2 stress 0.1495791
## Run 3 stress 0.1617926
## Run 4 stress 0.1231453
## ... New best solution
## ... Procrustes: rmse 1.896923e-05 max resid 7.76958e-05
## ... Similar to previous best
## Run 5 stress 0.1396627
## Run 6 stress 0.1617926
## Run 7 stress 0.1495181
## Run 8 stress 0.1770247
## Run 9 stress 0.1664312
## Run 10 stress 0.149518
## Run 11 stress 0.1537061
## Run 12 stress 0.1293893
## Run 13 stress 0.1770247
## Run 14 stress 0.1375489
## Run 15 stress 0.1365775
## Run 16 stress 0.1231453
## ... New best solution
## ... Procrustes: rmse 6.88286e-06 max resid 2.805843e-05
## ... Similar to previous best
## Run 17 stress 0.1665743
## Run 18 stress 0.1375489
## Run 19 stress 0.1375489
## Run 20 stress 0.1231453
## ... Procrustes: rmse 5.078507e-06 max resid 1.95447e-05
## ... Similar to previous best
## *** Best solution repeated 2 times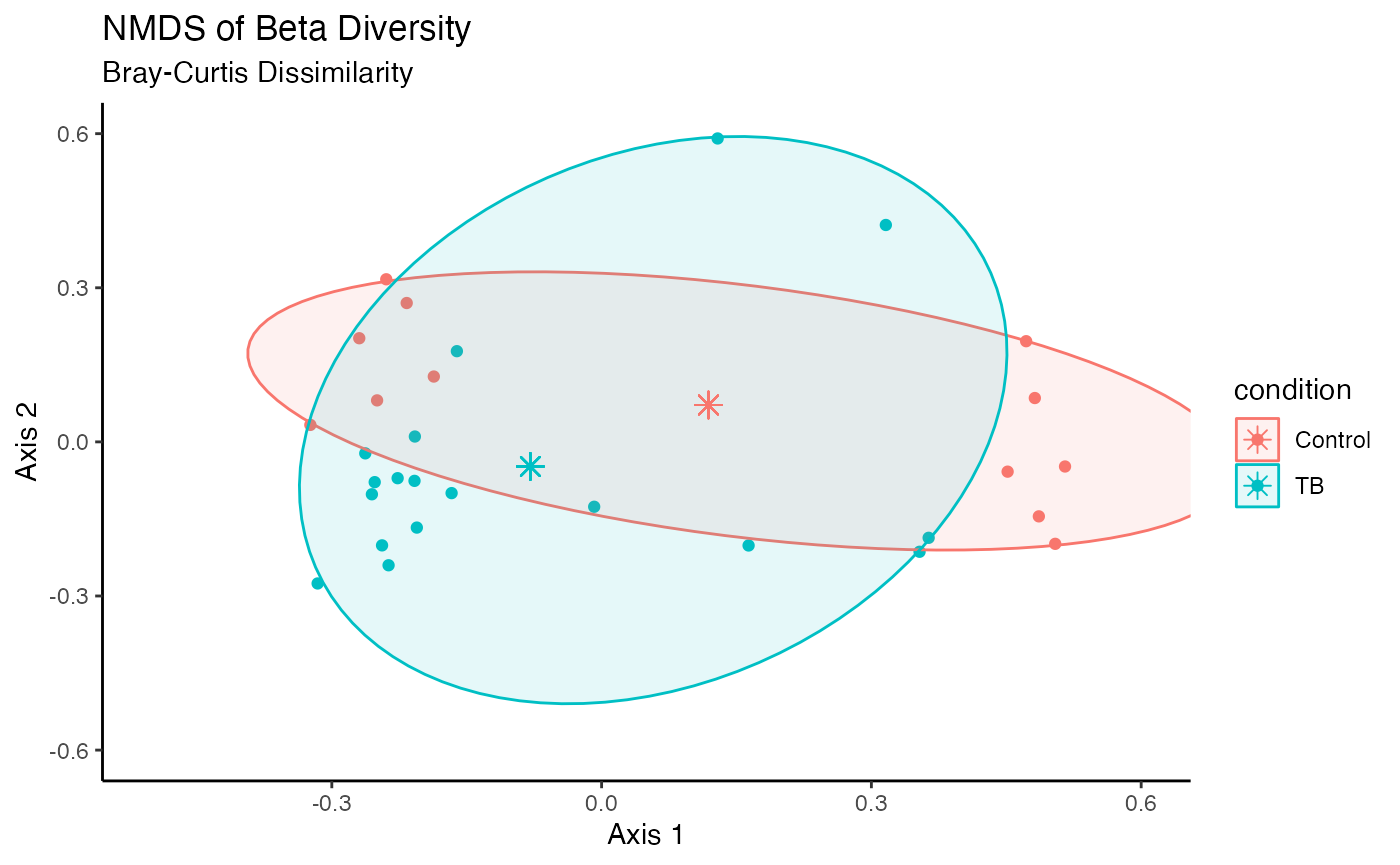
Dimensionality Reduction
PCA
A wrapper for conduction 2D and 3D Principal Component Analysis.
result <- dimred_pca(MAE,
tax_level = "genus",
color = "age_s",
shape = "Disease",
pcx = 1,
pcy = 2,
datatype = "logcpm"
)
result$plot
head(result$table)## PC Standard Deviation Variance Explained Cumulative Variance
## PC1 PC1 5.76 18.881% 18.88%
## PC2 PC2 4.54 11.732% 30.61%
## PC3 PC3 3.63 7.475% 38.09%
## PC4 PC4 3.53 7.066% 45.15%
## PC5 PC5 3.24 5.972% 51.12%
## PC6 PC6 2.95 4.943% 56.07%PCoA
A wrapper for conduction 2D and 3D Principal Coordinate Analysis.
result <- dimred_pcoa(MAE,
tax_level = "genus",
color = "age_s",
shape = "Disease",
axx = 1,
axy = 2,
method = "bray"
)
result$plot
head(result$table)## Axis Eigenvalue Variance Explained Cumulative Variance
## Axis.1 Axis.1 3.550 30.456% 30.46%
## Axis.2 Axis.2 1.590 14.187% 44.64%
## Axis.3 Axis.3 1.120 10.303% 54.95%
## Axis.4 Axis.4 0.751 7.224% 62.17%
## Axis.5 Axis.5 0.381 4.155% 66.32%
## Axis.6 Axis.6 0.304 3.521% 69.85%UMAP
A wrapper for conduction 2D and 3D Uniform Manifold Approximation & Projection.
result <- dimred_umap(MAE,
tax_level = "genus",
color = "age_s",
shape = "Disease",
cx = 1,
cy = 2,
n_neighbors = 15,
metric = "euclidean",
datatype = "logcpm"
)
result$plotDifferential Analysis
Here in animalcules, we provide a DESeq2-based differential abundance analysis. Users can choose the target variable, covariate variable, taxonomy level, minimum count cut-off, and an adjusted p-value threshold. The analysis report will output not only the adjusted p-value and log2-fold-change of the microbes, but also the percentage, prevalence, and the group size-adjusted fold change.
p <- differential_abundance(MAE,
tax_level = "phylum",
input_da_condition = c("Disease"),
min_num_filter = 2,
input_da_padj_cutoff = 0.5
)## estimating size factors## estimating dispersions## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimates## fitting model and testing## -- replacing outliers and refitting for 4 genes
## -- DESeq argument 'minReplicatesForReplace' = 7
## -- original counts are preserved in counts(dds)## estimating dispersions## fitting model and testing
p## microbe padj pValue log2FoldChange TB Control prevalence
## 1 Firmicutes 0.0936 0.0170 1.010 18/18 12/12 100.00%
## 2 Proteobacteria 0.0936 0.0109 1.970 17/18 12/12 96.67%
## 3 Bacteroidetes 0.1130 0.0307 -2.000 17/18 12/12 96.67%
## 4 Actinobacteria 0.1330 0.0484 -0.929 18/18 12/12 100.00%
## 5 Tenericutes 0.3350 0.1520 -2.610 5/18 3/12 26.67%
## 6 Cyanobacteria 0.4290 0.2340 -2.400 3/18 4/12 23.33%
## Group Size adjusted fold change
## 1 1.00
## 2 1.06
## 3 1.06
## 4 1.00
## 5 1.11
## 6 2.00Biomarker
One unique feature of animalcules is the biomarker identification module built on machine learning models. Users can choose one classification model from logistic regression, gradient boosting machine, or random forest to identify a microbes biomarker list. The feature importance score for each microbe will be provided. To evaluate the biomarker performance, the ROC plot and the AUC value using cross-validation outputs are shown to users. Note: Results may vary each run.
Train biomarker
p <- find_biomarker(MAE,
tax_level = "genus",
input_select_target_biomarker = c("Disease"),
nfolds = 3,
nrepeats = 3,
seed = 99,
percent_top_biomarker = 0.2,
model_name = "logistic regression"
)## Loading required package: ggplot2## Loading required package: lattice
# biomarker
p$biomarker## biomarker_list
## 1 Acidiphilium
## 2 Anaerostipes
## 3 Caulobacter
## 4 Corynebacterium
## 5 Filifactor
## 6 Geobacillus
## 7 Herbaspirillum
## 8 Mesorhizobium
## 9 Methylobacterium
## 10 Methylomonas
## 11 Moraxella
## 12 Mycobacterium
## 13 Novosphingobium
## 14 Paraburkholderia
## 15 Prevotella
## 16 Pseudarthrobacter
## 17 Pseudopropionibacterium
## 18 Rhodobacter
## 19 Sphingomonas
## 20 Streptococcus
## 21 Tannerella
# importance plot
p$importance_plot
# ROC plot
p$roc_plot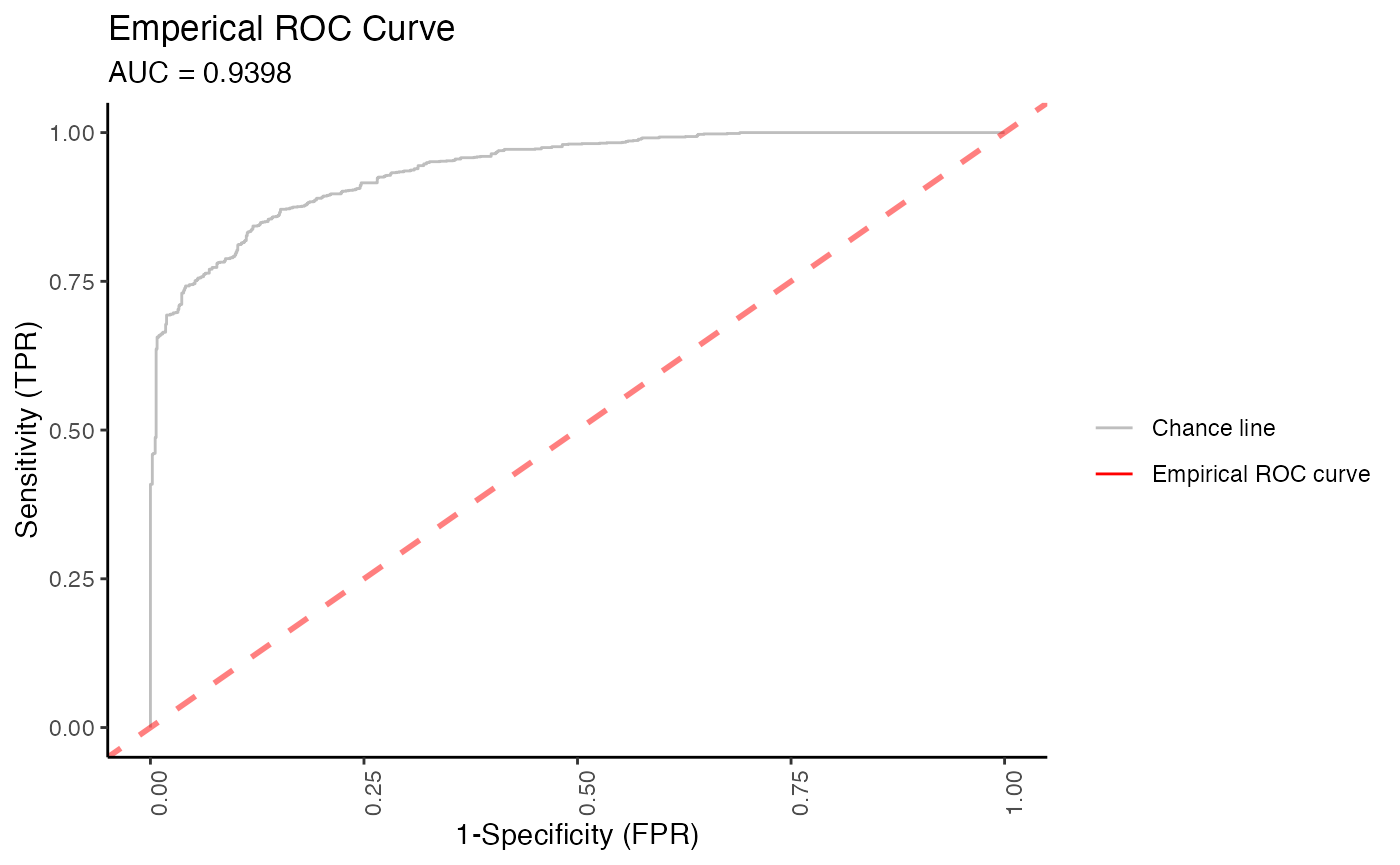
Session Info
## R version 4.3.2 (2023-10-31)
## Platform: aarch64-apple-darwin20 (64-bit)
## Running under: macOS Sonoma 14.2.1
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## time zone: America/New_York
## tzcode source: internal
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] caret_6.0-94 lattice_0.22-5
## [3] ggplot2_3.4.4 SummarizedExperiment_1.32.0
## [5] Biobase_2.62.0 GenomicRanges_1.54.1
## [7] GenomeInfoDb_1.38.5 IRanges_2.36.0
## [9] S4Vectors_0.40.2 BiocGenerics_0.48.1
## [11] MatrixGenerics_1.14.0 matrixStats_1.2.0
## [13] animalcules_1.19.2 BiocStyle_2.30.0
##
## loaded via a namespace (and not attached):
## [1] splines_4.3.2 bitops_1.0-7
## [3] tibble_3.2.1 polyclip_1.10-6
## [5] hardhat_1.3.0 pROC_1.18.5
## [7] XML_3.99-0.16 rpart_4.1.23
## [9] rex_1.2.1 lifecycle_1.0.4
## [11] globals_0.16.2 MASS_7.3-60.0.1
## [13] crosstalk_1.2.1 MultiAssayExperiment_1.28.0
## [15] magrittr_2.0.3 limma_3.58.1
## [17] plotly_4.10.4 sass_0.4.8
## [19] rmarkdown_2.25 jquerylib_0.1.4
## [21] yaml_2.3.8 ROCit_2.1.1
## [23] askpass_1.2.0 reticulate_1.34.0
## [25] RColorBrewer_1.1-3 lubridate_1.9.3
## [27] abind_1.4-5 zlibbioc_1.48.0
## [29] purrr_1.0.2 RCurl_1.98-1.14
## [31] nnet_7.3-19 tweenr_2.0.2
## [33] ipred_0.9-14 lava_1.7.3
## [35] GenomeInfoDbData_1.2.11 ggrepel_0.9.5
## [37] rmutil_1.1.10 inline_0.3.19
## [39] listenv_0.9.0 rentrez_1.2.3
## [41] vegan_2.6-4 umap_0.2.10.0
## [43] RSpectra_0.16-1 spatial_7.3-17
## [45] parallelly_1.36.0 pkgdown_2.0.7
## [47] permute_0.9-7 codetools_0.2-19
## [49] DelayedArray_0.28.0 DT_0.31
## [51] ggforce_0.4.1 shape_1.4.6
## [53] tidyselect_1.2.0 farver_2.1.1
## [55] stable_1.1.6 jsonlite_1.8.8
## [57] ellipsis_0.3.2 survival_3.5-7
## [59] iterators_1.0.14 systemfonts_1.0.5
## [61] foreach_1.5.2 tools_4.3.2
## [63] ragg_1.2.7 Rcpp_1.0.12
## [65] glue_1.7.0 prodlim_2023.08.28
## [67] SparseArray_1.2.3 BiocBaseUtils_1.4.0
## [69] xfun_0.41 mgcv_1.9-1
## [71] DESeq2_1.42.0 dplyr_1.1.4
## [73] withr_3.0.0 BiocManager_1.30.22
## [75] timeSeries_4032.109 fastmap_1.1.1
## [77] fansi_1.0.6 shinyjs_2.1.0
## [79] openssl_2.1.1 digest_0.6.34
## [81] timechange_0.2.0 R6_2.5.1
## [83] textshaping_0.3.7 colorspace_2.1-0
## [85] modeest_2.4.0 utf8_1.2.4
## [87] tidyr_1.3.0 generics_0.1.3
## [89] data.table_1.14.10 recipes_1.0.9
## [91] class_7.3-22 httr_1.4.7
## [93] htmlwidgets_1.6.4 S4Arrays_1.2.0
## [95] ModelMetrics_1.2.2.2 pkgconfig_2.0.3
## [97] gtable_0.3.4 tsne_0.1-3.1
## [99] timeDate_4032.109 covr_3.6.4
## [101] XVector_0.42.0 htmltools_0.5.7
## [103] bookdown_0.37 clue_0.3-65
## [105] scales_1.3.0 png_0.1-8
## [107] gower_1.0.1 knitr_1.45
## [109] rstudioapi_0.15.0 reshape2_1.4.4
## [111] statip_0.2.3 nlme_3.1-164
## [113] cachem_1.0.8 stringr_1.5.1
## [115] parallel_4.3.2 fBasics_4032.96
## [117] desc_1.4.3 pillar_1.9.0
## [119] grid_4.3.2 vctrs_0.6.5
## [121] cluster_2.1.6 evaluate_0.23
## [123] cli_3.6.2 locfit_1.5-9.8
## [125] compiler_4.3.2 rlang_1.1.3
## [127] crayon_1.5.2 future.apply_1.11.1
## [129] labeling_0.4.3 forcats_1.0.0
## [131] plyr_1.8.9 fs_1.6.3
## [133] stringi_1.8.3 viridisLite_0.4.2
## [135] BiocParallel_1.36.0 assertthat_0.2.1
## [137] munsell_0.5.0 lazyeval_0.2.2
## [139] glmnet_4.1-8 Matrix_1.6-5
## [141] stabledist_0.7-1 future_1.33.1
## [143] statmod_1.5.0 highr_0.10
## [145] GUniFrac_1.8 memoise_2.0.1
## [147] bslib_0.6.1 ape_5.7-1