Condensed MetaScope Tutorial
Aubrey Odom
Program in Bioinformatics, Boston University, Boston, MAaodom@bu.edu
October 30, 2024
Source:vignettes/MetaScopeTutorial.Rmd
MetaScopeTutorial.RmdThis is a condensed tutorial for the MetaScope package. We will walk through the individual steps to perform an analysis. A thorough explanation of the MetaScope package is available in the MetaScope Vignette.
Overview
The following diagram reveals the general module workflow of the MetaScope package.
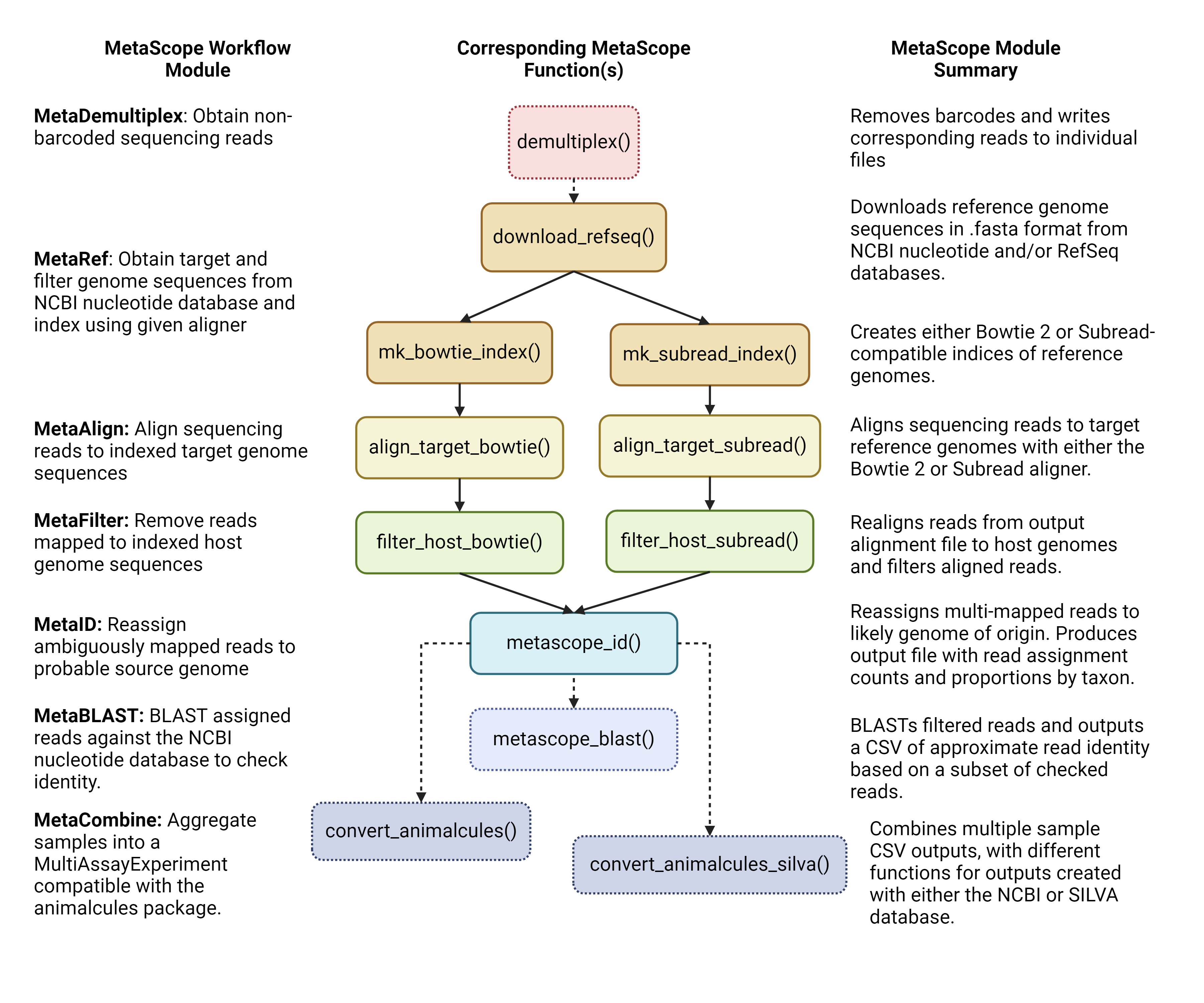
The core modules are as follows: 1. MetaDemultiplex:
Obtain non-barcoded sequencing reads 2. MetaRef: Obtain
target and filter genome sequences from NCBI nucleotide database and
index using a given aligner 3. MetaAlign: Align
sequencing reads to indexed target genome sequences 4.
MetaFilter: Remove reads mapped to indexed host genome
sequences 5. MetaID: Reassign ambiguously mapped reads
to probable source genome 6. MetaBLAST: BLAST assigned
reads against the NCBI nucleotide database to check identity 7.
MetaCombine: Aggregate samples into a
MultiAssayExperiment compatible with the animalcules R
package.
There are two sub-workflows that are included in the package, as seen in Figure 1: the Rbowtie2 and the Rsubread workflow. The major difference is that the functions in the MetaRef, MetaAlign, and MetaFilter modules differ by the aligner utilized.
Installation
In order to install MetaScope from GitHub, run the following code:
if (!requireNamespace("devtools", quietly = TRUE))
install.packages("devtools")
devtools::install_github("wejlab/MetaScope")Entrez key
We highly recommend grabbing an Entrez key using the function
taxize::use_entrez(). Then set your key as a global
environment variable like so:
NCBI_key <- "<Your key here>"
options("ENTREZ_KEY" = NCBI_key)MetaDemultiplex (optional)
This is an optional step in the analysis process depending on whether your reads are multiplexed. The reads which we are currently trying to analyze are not multiplexed and therefore this step is skipped in our analysis. The example shown below is using different reads that are barcoded in order to show the utility of the function.
Running meta_demultiplex
You will need the following files: 1. barcodeFile:
Path to a file containing a .tsv matrix with a header row, and then
sample names (column 1) and barcodes (column 2).
(barcodePath) 2. indexFile: Path to a
.fastq file that contains the barcodes for each read. The headers should
be the same (and in the same order) as readFile, and the sequence in the
indexFile should be the corresponding barcode for each read. Quality
scores are not considered. (indexPath) 3.
readFile: Path to the sequencing read .fastq file that
corresponds to the indexFile.
Additional parameters can be examined by accessing the
meta_demultiplex documentation with
?meta_demultiplex.
# Get barcode, index, and read data locations
barcodePath <-
system.file("extdata", "barcodes.txt", package = "MetaScope")
indexPath <- system.file("extdata", "virus_example_index.fastq",
package = "MetaScope")
readPath <-
system.file("extdata", "virus_example.fastq", package = "MetaScope")
# Get barcode, index, and read data locations
demult <-
meta_demultiplex(barcodePath,
indexPath,
readPath,
rcBarcodes = FALSE,
hammingDist = 2,
location = tempfile())
demultOutput
The meta_demultiplex function will returns multiple
.fastq files that contain all reads whose index matches the barcodes
given. These files will be written to the location directory, and will
be named based on the given sampleNames and barcodes,
e.g. ‘./demultiplex_fastq/SampleName1_GGAATTATCGGT.fastq.gz’
MetaRef: Reference Genome Library
Downloading NCBI nucleotide genomes
Most users will need to download a library of reference genomes if
they are not using their own library. This can be completed using the
download_refseq function. The following code chunks are not
run as they are quite large. This step is best completed by running a
background computing job as it may take some time.
Reference versus Representive genomes
The download_refseq function allows users to download
reference genomes from NCBI’s Reference Sequence (RefSeq) database and
the larger nucleotide database if desired. Reference genomes are defined
by the NCBI as high-quality, well-annotated genome sequence that serves
as a representative example for a particular species or organism.
A complete rundown of the differentiation between these categories is described on the NCBI website: https://www.ncbi.nlm.nih.gov/refseq/about/prokaryotes/
16S Analyses
For a 16S amplicon sequencing microbiome analysis, you will only need bacterial genomes. You can download all bacterial genomes from the NCBI RefSeq database like so:
# For RefSeq data, set reference = TRUE and representative = FALSE
download_refseq('bacteria', reference = TRUE, representative = FALSE,
out_dir = NULL, compress = TRUE, patho_out = FALSE,
caching = TRUE)
# If the RefSeq assortment is too limited, set representative = TRUE
download_refseq('bacteria', reference = TRUE, representative = FALSE,
out_dir = NULL, compress = TRUE, patho_out = FALSE,
caching = TRUE)Metagenomics / Whole Genome Sequencing
Target genomes
If you are performing a metagenomics analysis, you will likely want to analyze fungi, bacteria, and viruses. The example below is assuming that only reference genomes will be downloaded
for (taxa in c('bacteria', 'viruses', 'fungi')) {
download_refseq(taxa, reference = TRUE, representative = FALSE,
out_dir = NULL, compress = TRUE, patho_out = FALSE,
caching = TRUE)
}Host genomes
Given our ‘target’ genomes of interest in a metagenomics analysis we likely want to filter out any reads belonging to the host on which the sample was taken. So, in the instance that your host is human, you can download the reference genome using:
download_refseq("Homo sapiens", reference = TRUE, representative = FALSE,
out_dir = NULL, compress = TRUE, patho_out = FALSE,
caching = TRUE)Handpicked Reference Libraries
If you want to only use specific genomes in your libraries, you
should first check the included taxonomy table to check that your
organisms of interest are accessible. The taxonomy table object is
stored as MetaScope:::taxonomy_table. To search for your
organisms of interest, you can use any standard technique that you would
utilize to search for a table entry. For example, you can
useView(MetaScope:::taxonomy_table) and filter the table in
RStudio, or some other means of filtering the data.
The taxonomy table has the following structure:
head(MetaScope:::taxonomy_table)If I wanted to use only Staphylococcus bacterial genomes in this table then I could filter the taxonomy table like so:
all_staph_strains <- MetaScope:::taxonomy_table |>
dplyr::filter(genus == "Staphylococcus") |>
dplyr::pull(strain)
# Download representative genomes
sapply(all_staph_strains,
function(strain) download_refseq(strain, reference = TRUE, representative = TRUE,
out_dir = NULL, compress = TRUE, patho_out = FALSE,
caching = TRUE))Providing Your Own Reference Library
Databases outside of the NCBI nucleotide database are acceptable inputs as long as they are presented in .fasta format; the SILVA database is one such example. Please see the MetaScope tutorial, [Using the SILVA 16S rRNA Database with MetaScope][https://wejlab.github.io/metascope-docs/articles/SILVA.html].
Creating indices
Given your host and filter genomes, the next step is to create Bowtie- or Subread-compatible indices for the alignment modules.
Bowtie 2 indices
Be sure to indicate the folder location of your indices
(ref_dir). The folder where your indices should be output
is given to the lib_dir parameter. Finally, the library
name of your indices (the individual files rather than folder locations)
is given to lib_name. The number of parallel computing
threads is given to threads.
nThreads <- 1
# Example - target genome fungi
mk_bowtie_index(ref_dir = "fungi", lib_dir = "all_indices",
lib_name = "fungi", threads = nThreads, overwrite = TRUE)In the case that there are unresolved errors occurring unrelated to finding files or folders, we recommend troubleshooting by
- Adding the parameter
bowtie2_build_options = "--large-index" - Using the commandline Bowtie index creation tool,
Bowtie2-build
MetaAlign
We next perform the alignment step. Target genomes from reference libraries will now be aligned to sample reads to create a BAM file.
# If unpaired, see documentation
readPath1 <- "reads1.paired.fastq.gz"
readPath2 <- "reads2.paired.fastq.gz"
# Change to your target library names
targets <- c("fungi", "bacteria", "viruses")
alignment_directory <- "<where should alignments be output?>"
threads <- 8
# Usually expTag is a dynamically changing sample name
expTag <- "sampleX"
# Location of alignment indices
all_indices <- "<Location of alignment indices>"Bowtie 2
data("bt2_params")
# Use 16S params instead if you have 16S data
data("bt2_16S_params")
target_map <- align_target_bowtie(read1 = readPath1,
read2 = readPath2,
# Where are indices stored?
lib_dir = all_indices,
libs = targets,
align_dir = alignment_directory,
align_file = expTag,
overwrite = TRUE,
threads = threads,
# Use 16S params if you have 16S data
bowtie2_options = bt2_params,
quiet = FALSE)Subread
data("align_details")
target_map <- align_target(read1 = readPath1,
read2 = readPath2,
lib_dir = all_indices,
libs = targets,
threads = threads,
lib_dir = target_ref_temp,
align_file =
file.path(alignment_directory, expTag),
subread_options = align_details,
quiet = FALSE)MetaFilter
We will now filter out host reads using the host reference genome indices.
16S Samples
When processing a 16S rRNA amplicon sequencing sample, the MetaFilter step can be skipped entirely. You may proceed to the MetaID module.
Metagenomics Samples
For either Bowtie 2 or Subread alignments, the primary input file will be the alignment output by the MetaAlign module.
# Change to your filter library name(s)
filters <- c("homo_sapiens")
threads <- 8
output <- paste(paste0(alignment_directory, expTag), "filtered", sep = ".")Bowtie 2
data("bt2_params")
# target_map is the bam file output by the MetaAlign module
final_map <- filter_host_bowtie(reads_bam = target_map,
lib_dir = all_indices,
libs = filters,
make_bam = FALSE,
output = output,
threads = threads,
bowtie2_options = bt2_params,
overwrite = TRUE,
quiet = FALSE,
bowtie2_options = bt2_params)Subread
data("align_details")
final_map <- filter_host(
reads_bam = target_map,
lib_dir = all_indices,
make_bam = FALSE,
libs = filters,
output = output,
threads = threads,
subread_options = align_details)MetaID
The final step is to reassign ambiguously mapped reads to their likely source genome. This step is highly dependent on your reference library used, so please check the documentation or the SILVA tutorial if you used a library other than RefSeq or NCBI nucleotide.
Note that we highly recommend creating your own ENTREZ key, as noted earlier in this tutorial.
Bowtie 2
your_ENTREZ_KEY <- "<Your key here>"
outDir <- "<Your output directory>"
Sys.setenv(ENTREZ_KEY = your_ENTREZ_KEY)16S Sample
# NOTE: If 16S sample, use MetaAlign output
input_file <- target_map
metascope_id(input_file,
input_type = "bam",
aligner = "bowtie2",
NCBI_key = your_ENTREZ_KEY,
num_species_plot = 15,
quiet = FALSE,
out_dir = outDir)Metagenomics Sample
# NOTE: If Metagenomics sample, use MetaFilter output
input_file <- final_map
metascope_id(input_file,
input_type = "csv.gz",
aligner = "bowtie2",
NCBI_key = your_ENTREZ_KEY,
num_species_plot = 15,
quiet = FALSE,
out_dir = outDir)Subread
16S Sample
# NOTE: If 16S sample, use MetaAlign output
input_file <- target_map
metascope_id(input_file,
input_type = "bam",
aligner = "subread",
NCBI_key = your_ENTREZ_KEY,
num_species_plot = 15,
quiet = FALSE,
out_dir = outDir)Metagenomics sample
# NOTE: If Metagenomics sample, use MetaFilter output
input_file <- final_map
metascope_id(input_file,
input_type = "csv.gz",
aligner = "subread",
NCBI_key = your_ENTREZ_KEY,
num_species_plot = 15,
quiet = FALSE,
out_dir = outDir)MetaCombine
Assuming that you’ve processed several samples using MetaScope, you can aggregate all of your samples using the MetaCombine module. In this example, we assume our reference library was created with the MetaRef module (NCBI-formatted). This module produces a Multi-Assay Experiment
Create fake samples
tempfolder <- tempfile()
dir.create(tempfolder)
# Create three different samples
samp_names <- c("X123", "X456", "X789")
all_files <- file.path(tempfolder,
paste0(samp_names, ".csv"))
create_IDcsv <- function (out_file) {
final_taxids <- c("273036", "418127", "11234")
final_genomes <- c(
"Staphylococcus aureus RF122, complete sequence",
"Staphylococcus aureus subsp. aureus Mu3, complete sequence",
"Measles virus, complete genome")
best_hit <- sample(seq(100, 1050), 3)
proportion <- best_hit/sum(best_hit) |> round(2)
EMreads <- best_hit + round(runif(3), 1)
EMprop <- proportion + 0.003
dplyr::tibble(TaxonomyID = final_taxids,
Genome = final_genomes,
read_count = best_hit, Proportion = proportion,
EMreads = EMreads, EMProportion = EMprop) |>
dplyr::arrange(dplyr::desc(.data$read_count)) |>
utils::write.csv(file = out_file, row.names = FALSE)
message("Done!")
return(out_file)
}
out_files <- vapply(all_files, create_IDcsv, FUN.VALUE = character(1))Convert samples to MAE
outMAE <- convert_animalcules(meta_counts = out_files,
annot_path = annot_dat,
which_annot_col = "Sample",
end_string = ".metascope_id.csv",
qiime_biom_out = FALSE,
NCBI_key = NULL)
unlink(tempfolder, recursive = TRUE)Session Info
## R version 4.4.0 (2024-04-24)
## Platform: x86_64-pc-linux-gnu
## Running under: AlmaLinux 8.10 (Cerulean Leopard)
##
## Matrix products: default
## BLAS/LAPACK: FlexiBLAS NETLIB; LAPACK version 3.11.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: America/New_York
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] BiocStyle_2.32.1
##
## loaded via a namespace (and not attached):
## [1] vctrs_0.6.5 cli_3.6.3 knitr_1.48
## [4] rlang_1.1.4 xfun_0.45 purrr_1.0.2
## [7] textshaping_0.3.7 jsonlite_1.8.9 htmltools_0.5.8.1
## [10] ragg_1.3.0 sass_0.4.9 rmarkdown_2.28
## [13] evaluate_1.0.0 jquerylib_0.1.4 fastmap_1.2.0
## [16] yaml_2.3.10 lifecycle_1.0.4 memoise_2.0.1
## [19] bookdown_0.39 BiocManager_1.30.22 compiler_4.4.0
## [22] fs_1.6.4 htmlwidgets_1.6.4 rstudioapi_0.16.0
## [25] systemfonts_1.0.6 digest_0.6.37 R6_2.5.1
## [28] magrittr_2.0.3 bslib_0.8.0 tools_4.4.0
## [31] pkgdown_2.0.9 cachem_1.1.0 desc_1.4.3