Introduction to MetaScope
Aubrey Odom
Program in Bioinformatics, Boston University, Boston, MAaodom@bu.edu
Rahul Varki
Research Assistant in Johnson Lab, Boston University School of Medicine, Boston, MArvarki@bu.edu
W. Evan Johnson
The Section of Computational Biomedicine, Boston University School of Medicine, Boston, MAwej@bu.edu
April 25, 2025
Source:vignettes/MetaScope_vignette.Rmd
MetaScope_vignette.RmdImportant Note
Although it is not a requirement, MetaScope will fun faster and more efficiently for larger samples if the samtools package is present on your device. You can download it at https://github.com/samtools/samtools.
Introduction
MetaScope is a complete metagenomics profiling package that can accurately identify the composition of microbes within a sample at a strain-level resolution. MetaScope can be considered as an updated and expanded R translation of PathoScope 2.0, a Python-based metagenomic profiling package created by the Johnson lab. A few improvements made in MetaScope include using the BAM file format instead of the SAM file format for significantly less disk space usage, removing all dependencies to NCBI’s now defunct GI sequence annotations, and properly filtering reads that align to filter reference genomes. Functions to analyze host microbiome data are also planned to be added in future updates to the package.
MetaScope Workflows
The major workflow of MetaScope is delineated below. It is composed of core modules that are formed by groups of functions.
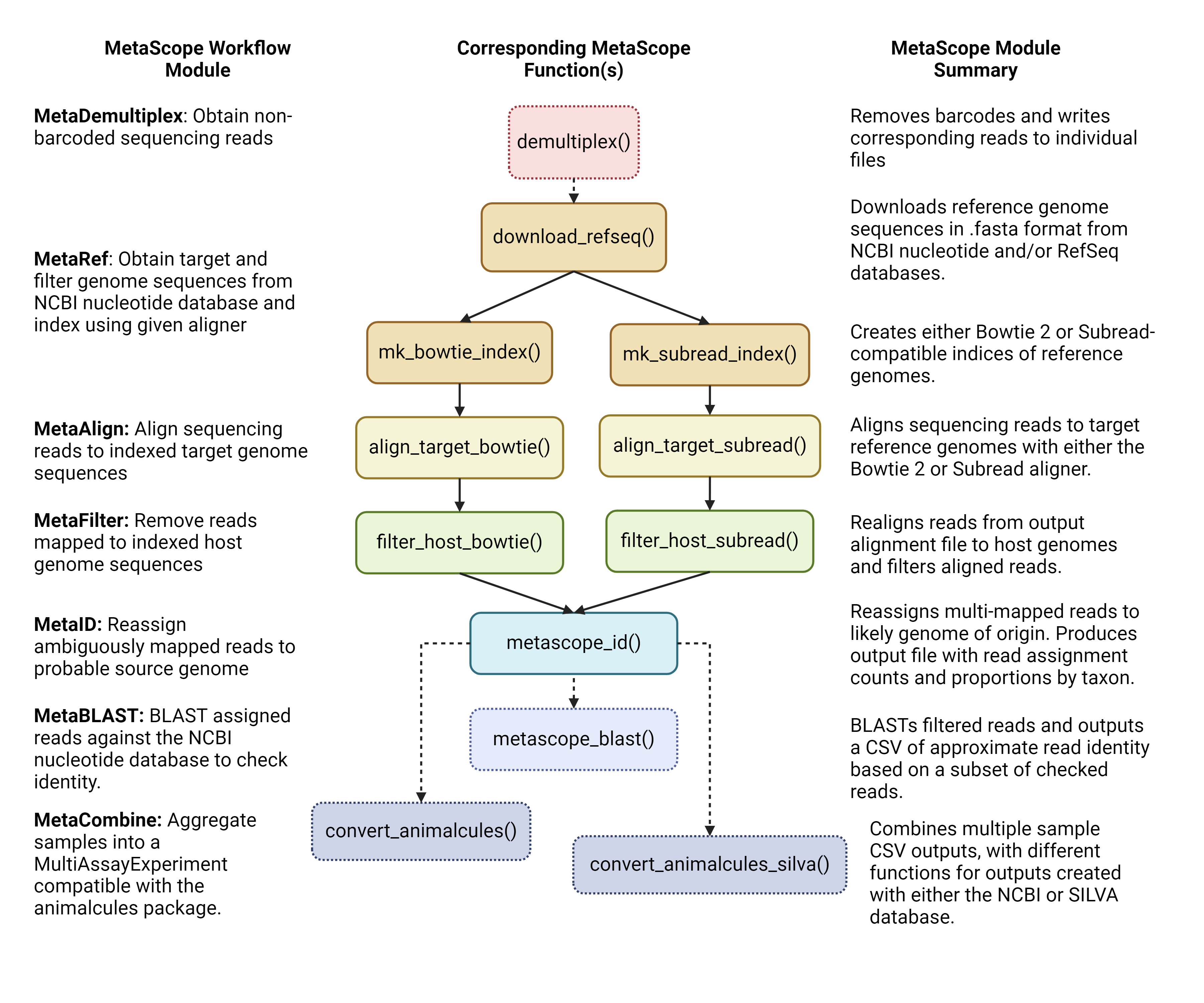
The core modules are as follows: 1. MetaDemultiplex:
Obtain non-barcoded sequencing reads 2. MetaRef: Obtain
target and filter genome sequences from NCBI nucleotide database and
index using a given aligner 3. MetaAlign: Align
sequencing reads to indexed target genome sequences 4.
MetaFilter: Remove reads mapped to indexed host genome
sequences 5. MetaID: Reassign ambiguously mapped reads
to probable source genome 6. MetaCombine: Aggregate
samples into a MultiAssayExperiment compatible with the
animalcules R package.
There are two sub-workflows that are included in the package, as seen in Figure 1: the Rbowtie2 and the Rsubread workflow. The major difference is that the functions in the MetaRef, MetaAlign, and MetaFilter modules differ by the aligner utilized.
The Rbowtie2 functions utilize the Bowtie2 aligner
(Langmead 2012) whereas the Rsubread
functions utilize the Rsubread aligner (Liao 2019). The nuances of how
to use each function can be found by looking at each function’s help
manual (R command: ?<name of function>). For
reference, PathoScope 2.0 uses the Bowtie2 aligner in its workflow.
In this vignette, we will analyze the mock data provided in the package via the Rbowtie2 sub-workflow. We will utilize all of the core modules in sequential order. We will make mention of the equivalent Rsubread function whenever an Rbowtie2 function is being used. For the purposes of this example, the MetaCombine module will be omitted.
Installation
In order to install MetaScope from Bioconductor, run the following code:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("MetaScope")Data
The mock data provided in the package consists of simulated
sequencing data generated from the SAMtools
wgsim function (see extdata_explanations.Rmd
in inst/script to see exact commands). The
wgsim function is a tool which allows for the generation of
FASTQ reads from a reference genome (FASTA). The mock data
(reads.fastq) contains 1500 reads, of which 1000 reads are
derived from the Staphylococcus aureus RF122 strain and 500
reads are derived from the Staphylococcus epidermidis RP62A
strain. In this data set, we assume that the S. aureus RF122
reads are the reads of interest, and the S. epidermidis RP62A
reads are known contaminant reads which should be removed during the
analysis. Ideally, the microbial composition report (.csv) produced at
the end of the analysis should contain only reads assigned to the S.
aureus RF122 strain.
MetaDemultiplex: Demultiplexing reads
Sequence runs on NGS instruments are typically carried out with
multiple samples pooled together. An index tag (also called a barcode)
consisting of a unique sequence of between 6 and 12bp is added to each
sample so that the sequence reads from different samples can be
identified. For 16s experiments or sequencing conducted using an
Illumina machine, the process of demultiplexing (dividing your sequence
reads into separate files for each index tag/sample) and generating the
FASTQ data files required for downstream analysis can be done using the
MetaScope demultiplexing workflow. This consists of the
meta_demultiplex() function, which takes as arguments a
matrix of sample names/barcodes, a FASTQ file of barcodes by sequence
header, and a FASTQ file of reads corresponding to the barcodes. Based
on the barcodes given, the function extracts all reads for the indexed
barcode and writes all the reads from that barcode to separate FASTQ
files.
This is an optional step in the analysis process depending on whether your reads are multiplexed. The reads which we are currently trying to analyze are not multiplexed and therefore this step is skipped in our analysis. The example shown below is using different reads that are barcoded in order to show the utility of the function.
# Get barcode, index, and read data locations
barcodePath <-
system.file("extdata", "barcodes.txt", package = "MetaScope")
indexPath <- system.file("extdata", "virus_example_index.fastq",
package = "MetaScope")
readPath <-
system.file("extdata", "virus_example.fastq", package = "MetaScope")
# Get barcode, index, and read data locations
demult <-
meta_demultiplex(barcodePath,
indexPath,
readPath,
rcBarcodes = FALSE,
hammingDist = 2,
location = tempfile())
demult## SampleName Barcode NumberOfReads
## 1 CDV TCCACGT 25
## 2 LaCrosse ACAGGCT 25
## 3 RSV ATCGTGC 25
## 4 EboV ACTACAG 25
## 5 Measles AAGTCGC 25
## 6 VSV TCTCAGG 25MetaRef: Reference Genome Library
The MetaScope genome library workflow is designed to assist with collection of sequence data from the National Center for Biotechnology Information (NCBI) nucleotide database. Prior to doing so, the potential targets and filters for the analysis should be identified. That is, what “target” species do you expect to find in your metagenomic sample that you would like to identify, and what reads would you like to “filter” out from the data that are not essential to your analysis?
Typically, the targets of the analysis are microbes (that is, viruses, bacteria, and fungi), and we wish to filter out or discard any reads from the host in addition to artificially added sequences, such as Phi X 174. Following identification of the targets and filters, we use a reference genome library to align the vast number of sample reads back to the respective regions of origin in various species.
The download_refseq() function automatically extracts
custom reference genome libraries in a FASTA file for microbial or host
genomes. The user must first indicate a taxon for which to download the
genomes, such as ‘bacteria’ or ‘primates’. A table of possible entries
can be viewed by accessing the MetaScope:::taxonomy_table
object. The user may then specify whether they wish to download only the
RefSeq reference genomes, or both the reference and representative
genomes. The compress option then allows users to specify whether to
compress the output FASTA file; this is done by default.
Creating a Taxonomy Database
A reference of taxonomy reference identifiers (e.g. NCBI accessions)
is critical for fully identifying microbes with the MetaScope pipeline.
This step should be completed ONCE, PRIOR TO RUNNING ANY AND ALL SAMPLES
with the download_accessions() function. For an example of
how to make a typical database for all host and filter genomes in your
sample, please see the example
script on our website.
If using the SILVA 16S database, NCBI nucleotide or RefSeq database,
you can use download_accessions() with the following
arguments:
download_accessions(
ind_dir = "C:/Users/JohnSmith/ResearchIndices",
tmp_dir = file_path(ind_dir, "tmp"),
remove_tmp_dir = TRUE,
# For NCBI RefSeq or nucleotide database
NCBI_accessions_database = TRUE,
NCBI_accessions_name = "accessionTaxa.sql",
# For SILVA 16S database
silva_taxonomy_database = TRUE,
silva_taxonomy_name = "all_silva_headers.rds"
)However, for this vignette, we know in advance precisely which
genomes and strains we will be testing. Therefore, we will generate a
small subdatabase of taxonomizr references. We do not need to use
download_accessions() in this case.
tmp_accession <- system.file("extdata","example_accessions.sql", package = "MetaScope")Downloading target genomes
Even though in this scenario we know exactly from where the reads in
the mock data (reads.fastq) originate, in most cases we may
only have a general idea of read origins. Therefore, in the following
code, we will download the genome of the Staphylococcus aureus
RF122 strain along with the genomes of a few other closely related
Staphylococcus aureus strains from the NCBI RefSeq database.
These genomes together will act as our target genome library.
target_ref_temp <- tempfile()
dir.create(target_ref_temp)
all_species <- c("Staphylococcus aureus subsp. aureus Mu50",
"Staphylococcus aureus subsp. aureus Mu3",
"Staphylococcus aureus subsp. aureus str. Newman",
"Staphylococcus aureus subsp. aureus N315",
"Staphylococcus aureus RF122",
"Staphylococcus aureus subsp. aureus ST398")
sapply(all_species, download_refseq,
reference = FALSE, representative = FALSE, compress = TRUE,
out_dir = target_ref_temp, caching = TRUE, accession_path = tmp_accession)## Staphylococcus aureus subsp. aureus Mu50
## "/scratch/339945.1.ood/RtmprC2Oqu/file1a6bba29554032/Staphylococcus_aureus_subsp._aureus_Mu50.fasta.gz"
## Staphylococcus aureus subsp. aureus Mu3
## "/scratch/339945.1.ood/RtmprC2Oqu/file1a6bba29554032/Staphylococcus_aureus_subsp._aureus_Mu3.fasta.gz"
## Staphylococcus aureus subsp. aureus str. Newman
## "/scratch/339945.1.ood/RtmprC2Oqu/file1a6bba29554032/Staphylococcus_aureus_subsp._aureus_str._Newman.fasta.gz"
## Staphylococcus aureus subsp. aureus N315
## "/scratch/339945.1.ood/RtmprC2Oqu/file1a6bba29554032/Staphylococcus_aureus_subsp._aureus_N315.fasta.gz"
## Staphylococcus aureus RF122
## "/scratch/339945.1.ood/RtmprC2Oqu/file1a6bba29554032/Staphylococcus_aureus_RF122.fasta.gz"
## Staphylococcus aureus subsp. aureus ST398
## "/scratch/339945.1.ood/RtmprC2Oqu/file1a6bba29554032/Staphylococcus_aureus_subsp._aureus_ST398.fasta.gz"Downloading filter genomes
We will also download the reference genome and related sequences* of the Staphylococcus epidermidis RP62A strain from the NCBI nucleotide database, in an uncompressed FASTA format. This genome will act as our filter library.
- since we are downloading from the nucleotide database with representative and reference = FALSE, several sequences will be downloaded in addition to the main genome.
filter_ref_temp <- tempfile()
dir.create(filter_ref_temp)
download_refseq(
taxon = "Staphylococcus epidermidis RP62A",
representative = FALSE, reference = FALSE,
compress = TRUE, out_dir = filter_ref_temp,
caching = TRUE,
accession_path = tmp_accession)## [1] "/scratch/339945.1.ood/RtmprC2Oqu/file1a6bba267b8dfb/Staphylococcus_epidermidis_RP62A.fasta.gz"Creating indices using a given aligner
We now use mk_bowtie_index(), a wrapper for the
Rbowtie2::bowtie2_build function, to generate Bowtie2
compatible indexes from the reference genomes that were previously
downloaded. The target and reference genome files (.fasta or .fasta.gz
extension) must be placed into their own separate empty directories
prior to using the function. This is due to the fact that the function
will attempt to build the indexes from all the files present in the
directory. The function will give an error if other files (other than
.fasta or .fasta.gz) are present in the directory. Depending on the
combined size of the reference genomes, the function will automatically
create either small (.bt2) or large (.bt2l) Bowtie2 indexes.
The target and filter reference genomes downloaded in the previous step have been combined and renamed to target.fasta and filter.fasta respectively for convenience. These are the files from which the Bowtie2 indexes will be made from.
# Create temp directory to store the Bowtie2 indices
index_temp <- tempfile()
dir.create(index_temp)
# Create target index
mk_bowtie_index(
ref_dir = target_ref_temp,
lib_dir = index_temp,
lib_name = "target",
overwrite = TRUE
)## arguments 'show.output.on.console', 'minimized' and 'invisible' are for Windows only## Index building complete## [1] "/scratch/339945.1.ood/RtmprC2Oqu/file1a6bba32650d70"
# Create filter index
mk_bowtie_index(
ref_dir = filter_ref_temp,
lib_dir = index_temp,
lib_name = "filter",
overwrite = TRUE
)## arguments 'show.output.on.console', 'minimized' and 'invisible' are for Windows only
## Index building complete## [1] "/scratch/339945.1.ood/RtmprC2Oqu/file1a6bba32650d70"Alignment with Reference Libraries
After acquiring the target and filter genome libraries, we then take the sequencing reads from our sample and map them first to the target library and then to the filter library. MetaScope’s Rbowtie2 mapping function utilizes the Bowtie2 aligner (Langmead 2012) which maps reads to a reference genome using a full-text minute index based approach. Essentially, the algorithm extracts substrings which are referred to as “seeds” from the reads and aligns them to the reference genomes with the assistance from the full-text minute index. Seed alignments to the reference genomes are prioritized and then finally extended into full alignments using dynamic programming.
MetaAlign
Following index creation, we will use the Bowtie2 aligner to map the
reads to the target genomes with the align_target_bowtie()
function (Rsubread equivalent: align_target()). The
function takes as an input the location of the FASTQ file to align, the
directory where the indexes are stored, the names of the indexes to
align against, the directory where the BAM file should be written, and
the basename of the output BAM file.
In practice, align_target_bowtie() maps reads to each
target library separately, removes the unmapped reads from each file,
and finally merges and sorts by chromosome the BAM files from each
library into a single output file (same with align_target).
If SAMtools is installed on the machine and can be found by
the Sys.which("samtools") R command, the BAM file will be
directly created, otherwise an intermediate SAM file will be created
prior to the creation of the BAM file which could potentially create
issues if the SAM file is large and there is limited disk space. The
default alignment parameters are the same as PathoScope 2.0’s default
alignment parameters, but users can provide their own Bowtie 2 alignment
settings if desired.
We will now align the sample reads (reads.fastq) to the target reference genomes using the Bowtie 2 indexes that we just built.
# Create a temp directory to store output bam file
output_temp <- tempfile()
dir.create(output_temp)
# Get path to example reads
readPath <-
system.file("extdata", "reads.fastq", package = "MetaScope")
# Align reads to the target genomes
target_map <-
align_target_bowtie(
read1 = readPath,
lib_dir = index_temp,
libs = "target",
align_dir = output_temp,
align_file = "bowtie_target",
overwrite = TRUE
)## [1] "Samtools found on system. Using samtools to create bam file"## arguments 'show.output.on.console', 'minimized' and 'invisible' are for Windows onlyMetaFilter
The last step in the mapping workflow is to filter the output BAM
file according to the reference genome for the filter/host species.
Although we have already filtered out any unmapped reads, which may
belong to one or more host species or otherwise, there may still remain
some sort of unwelcome contamination in the data from the filter species
which we wish to remove. To do this, we employ
filter_host_bowtie() (Rsubread equivalent:
filter_host()), which takes as an input the location of the
BAM file created from align_target_bowtie(), the directory
where the indexes are stored, and the names of the filter indexes to
align against, to produce a sorted BAM file with any reads that match
the filter libraries removed. We will then use this final BAM file
downstream for further analysis.
final_map <-
filter_host_bowtie(
reads_bam = target_map,
lib_dir = index_temp,
libs = "filter",
make_bam = TRUE, # Set to true to create BAM output
# Default is to create simplified .csv.gz output
# The .csv.gz output is much quicker to create!
overwrite = TRUE,
threads = 1
)## | | | 0% | |======================================================================| 100%
## [1] "Samtools found on system. Using samtools to create bam file"## arguments 'show.output.on.console', 'minimized' and 'invisible' are for Windows onlyEvaluating alignments (supplemental)
Note: the next two code blocks are included for the sake of examining the vignette example, but are not useful for “real life” data. To continue your analysis, head down to the Genome Identification header.
Prior to the last step in the analysis, we will look at the primary
alignments of the mapped reads in the filtered BAM file that we just
created using the filter_host_bowtie() function. According
to the Bowtie2
manual, a primary alignment is described as the alignment that
received the highest alignment score among all alignments for that read.
When looking at the primary alignments of the mapped reads, we can see
that the majority of reads have mapped to the correct Staphylococcus
aureus RF122 strain. However, some residual reads have primary
alignments to the other S. aureus strains which are incorrect.
If we were to stop the analysis at this point, we could potentially be
lead to believe that our sample has increased microbial diversity, when
it actually does not.
bamFile <- Rsamtools::BamFile(final_map)
param <-
Rsamtools::ScanBamParam(
flag = Rsamtools::scanBamFlag(isSecondaryAlignment = FALSE),
what = c("flag", "rname")
) #Gets info about primary alignments
aln <- Rsamtools::scanBam(bamFile, param = param)
accession_all <- aln[[1]]$rname
genome_name_all <- accession_all |>
taxonomizr::accessionToTaxa(tmp_accession) |>
taxonomizr::getTaxonomy(sqlFile = tmp_accession, desiredTaxa = "strain")
read_count_table <- sort(table(genome_name_all), decreasing = TRUE)
knitr::kable(
read_count_table,
col.names = c("Genome Assigned", "Read Count"))| Genome Assigned | Read Count |
|---|---|
| Staphylococcus aureus RF122 | 792 |
| Staphylococcus aureus subsp. aureus ST398 | 61 |
| Staphylococcus aureus subsp. aureus Mu3 | 53 |
| Staphylococcus aureus subsp. aureus str. Newman | 45 |
| Staphylococcus aureus subsp. aureus Mu50 | 42 |
We can also look at the secondary alignments of the mapped reads within our filtered BAM file. A secondary alignment occurs when a read maps to multiple different genomes. We can see that the majority of our secondary alignments are to the other Staphylococcus aureus strains, which makes sense considering that the majority of the primary alignments were to the correct Staphylococcus aureus RF122 strain.
bamFile <- Rsamtools::BamFile(final_map)
param <-
Rsamtools::ScanBamParam(
flag = Rsamtools::scanBamFlag(isSecondaryAlignment = TRUE),
what = c("flag", "rname")
) #Gets info about secondary alignments
aln <- Rsamtools::scanBam(bamFile, param = param)
accession_all <- aln[[1]]$rname
genome_name_all <- accession_all |>
taxonomizr::accessionToTaxa(tmp_accession) |>
taxonomizr::getTaxonomy(sqlFile = tmp_accession, desiredTaxa = "strain")
read_count_table <- sort(table(genome_name_all), decreasing = TRUE)
knitr::kable(
read_count_table,
col.names = c("Genome Assigned", "Read Count"))| Genome Assigned | Read Count |
|---|---|
| Staphylococcus aureus subsp. aureus ST398 | 1258 |
| Staphylococcus aureus subsp. aureus str. Newman | 686 |
| Staphylococcus aureus subsp. aureus Mu3 | 673 |
| Staphylococcus aureus subsp. aureus Mu50 | 451 |
| Staphylococcus aureus RF122 | 98 |
MetaID: Origin Genome Identification
Following the proper alignment of a sample to all target and filter libraries of interest, we may proceed in identifying which genomes are most likely to be represented in the sample. This identification workflow is the core of MetaScope; it features a Bayesian read reassignment model which dramatically improves specificity and sensitivity over other methods (Francis et. al 2013). This is because such a method identifies reads with unique alignments and uses them to guide the reassignment of reads with ambiguous alignments.
The MetaID identification module consists of a single function,
MetaScope_ID(), which reads in a .bam file, annotates the
taxonomy and genome names, reduces the mapping ambiguity using a mixture
model, and outputs a .csv file with the results. Currently, it assumes
that the genome library/.bam files use NCBI accession names for
reference names.
output <- metascope_id(
final_map,
input_type = "bam",
# change input_type to "csv.gz" when not creating a BAM
aligner = "bowtie2",
num_species_plot = 0,
accession_path = tmp_accession
)
knitr::kable(output,
format = "html",
digits = 2,
caption = "Table of MetaScope ID results")| TaxonomyID | Genome | read_count | Proportion | readsEM | EMProportion |
|---|---|---|---|---|---|
| 273036 | Bacteria;Bacillota;Bacilli;Bacillales;Staphylococcaceae;Staphylococcus;Staphylococcus aureus;Staphylococcus aureus RF122 | 855 | 0.84 | 851.9 | 0.84 |
| 418127 | Bacteria;Bacillota;Bacilli;Bacillales;Staphylococcaceae;Staphylococcus;Staphylococcus aureus;Staphylococcus aureus subsp. aureus Mu3 | 131 | 0.13 | 124.8 | 0.12 |
| 426430 | Bacteria;Bacillota;Bacilli;Bacillales;Staphylococcaceae;Staphylococcus;Staphylococcus aureus;Staphylococcus aureus subsp. aureus str. Newman | 30 | 0.03 | 39.0 | 0.04 |
| 523796 | Bacteria;Bacillota;Bacilli;Bacillales;Staphylococcaceae;Staphylococcus;Staphylococcus aureus;Staphylococcus aureus subsp. aureus ST398 | 3 | 0.00 | 3.2 | 0.00 |
| 158878 | Bacteria;Bacillota;Bacilli;Bacillales;Staphylococcaceae;Staphylococcus;Staphylococcus aureus;Staphylococcus aureus subsp. aureus Mu50 | 1 | 0.00 | 1.1 | 0.00 |
Note: the next code block is included for the sake of
examining the vignette example, but can be skipped. Your results are in
the CSV file produced by the metascope_id()
function.
We will now look at the read reassignment results reported in the output CSV file.
relevant_col <- dirname(final_map) %>%
file.path("bowtie_target.metascope_id.csv") %>%
read.csv() %>% dplyr::select(2:4)
relevant_col |>
dplyr::mutate(
Genome = stringr::str_replace_all(Genome, ',.*', ''),
Genome = stringr::str_replace_all(Genome, "(ST398).*", "\\1"),
Genome = stringr::str_replace_all(Genome, "(N315).*", "\\1"),
Genome = stringr::str_replace_all(Genome, "(Newman).*", "\\1"),
Genome = stringr::str_replace_all(Genome, "(Mu3).*", "\\1"),
Genome = stringr::str_replace_all(Genome, "(RF122).*", "\\1")
) |>
knitr::kable()| Genome | read_count | Proportion |
|---|---|---|
| Bacteria;Bacillota;Bacilli;Bacillales;Staphylococcaceae;Staphylococcus;Staphylococcus aureus;Staphylococcus aureus RF122 | 855 | 0.8382353 |
| Bacteria;Bacillota;Bacilli;Bacillales;Staphylococcaceae;Staphylococcus;Staphylococcus aureus;Staphylococcus aureus subsp. aureus Mu3 | 131 | 0.1284314 |
| Bacteria;Bacillota;Bacilli;Bacillales;Staphylococcaceae;Staphylococcus;Staphylococcus aureus;Staphylococcus aureus subsp. aureus str. Newman | 30 | 0.0294118 |
| Bacteria;Bacillota;Bacilli;Bacillales;Staphylococcaceae;Staphylococcus;Staphylococcus aureus;Staphylococcus aureus subsp. aureus ST398 | 3 | 0.0029412 |
| Bacteria;Bacillota;Bacilli;Bacillales;Staphylococcaceae;Staphylococcus;Staphylococcus aureus;Staphylococcus aureus subsp. aureus Mu50 | 1 | 0.0009804 |
unlink(".bowtie2.cerr.txt")We can see that the read reassignment function has reassigned the majority of the ambiguous alignments back to the Staphylococcus aureus RF122 strain, the correct strain of origin.
Session Info
## R version 4.4.3 (2025-02-28)
## Platform: x86_64-pc-linux-gnu
## Running under: AlmaLinux 8.10 (Cerulean Leopard)
##
## Matrix products: default
## BLAS/LAPACK: FlexiBLAS NETLIB; LAPACK version 3.11.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: America/New_York
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] magrittr_2.0.3 MetaScope_1.9.1 BiocStyle_2.34.0
##
## loaded via a namespace (and not attached):
## [1] xfun_0.52 bslib_0.9.0 htmlwidgets_1.6.4
## [4] lattice_0.22-6 vctrs_0.6.5 tools_4.4.3
## [7] bitops_1.0-9 generics_0.1.3 parallel_4.4.3
## [10] stats4_4.4.3 curl_6.2.2 tibble_3.2.1
## [13] RSQLite_2.3.9 blob_1.2.4 pkgconfig_2.0.3
## [16] Matrix_1.7-3 data.table_1.17.99 dbplyr_2.5.0
## [19] desc_1.4.3 S4Vectors_0.44.0 lifecycle_1.0.4
## [22] GenomeInfoDbData_1.2.13 compiler_4.4.3 stringr_1.5.1
## [25] Rsamtools_2.22.0 textshaping_1.0.0 Biostrings_2.74.1
## [28] codetools_0.2-20 GenomeInfoDb_1.42.3 htmltools_0.5.8.1
## [31] sass_0.4.9 yaml_2.3.10 pillar_1.10.2
## [34] pkgdown_2.1.1 crayon_1.5.3 jquerylib_0.1.4
## [37] BiocParallel_1.40.0 cachem_1.1.0 taxonomizr_0.11.1
## [40] tidyselect_1.2.1 digest_0.6.37 stringi_1.8.7
## [43] dplyr_1.1.4 purrr_1.0.4 bookdown_0.42
## [46] grid_4.4.3 fastmap_1.2.0 cli_3.6.5
## [49] withr_3.0.2 filelock_1.0.3 UCSC.utils_1.2.0
## [52] bit64_4.6.0-1 rmarkdown_2.29 XVector_0.46.0
## [55] httr_1.4.7 bit_4.6.0 ragg_1.3.3
## [58] memoise_2.0.1 evaluate_1.0.3 knitr_1.50
## [61] GenomicRanges_1.58.0 IRanges_2.40.1 BiocFileCache_2.14.0
## [64] rlang_1.1.6 glue_1.8.0 DBI_1.2.3
## [67] BiocManager_1.30.25 BiocGenerics_0.52.0 rstudioapi_0.17.1
## [70] jsonlite_2.0.0 Rbowtie2_2.12.3 R6_2.6.1
## [73] systemfonts_1.2.1 fs_1.6.5 zlibbioc_1.52.0