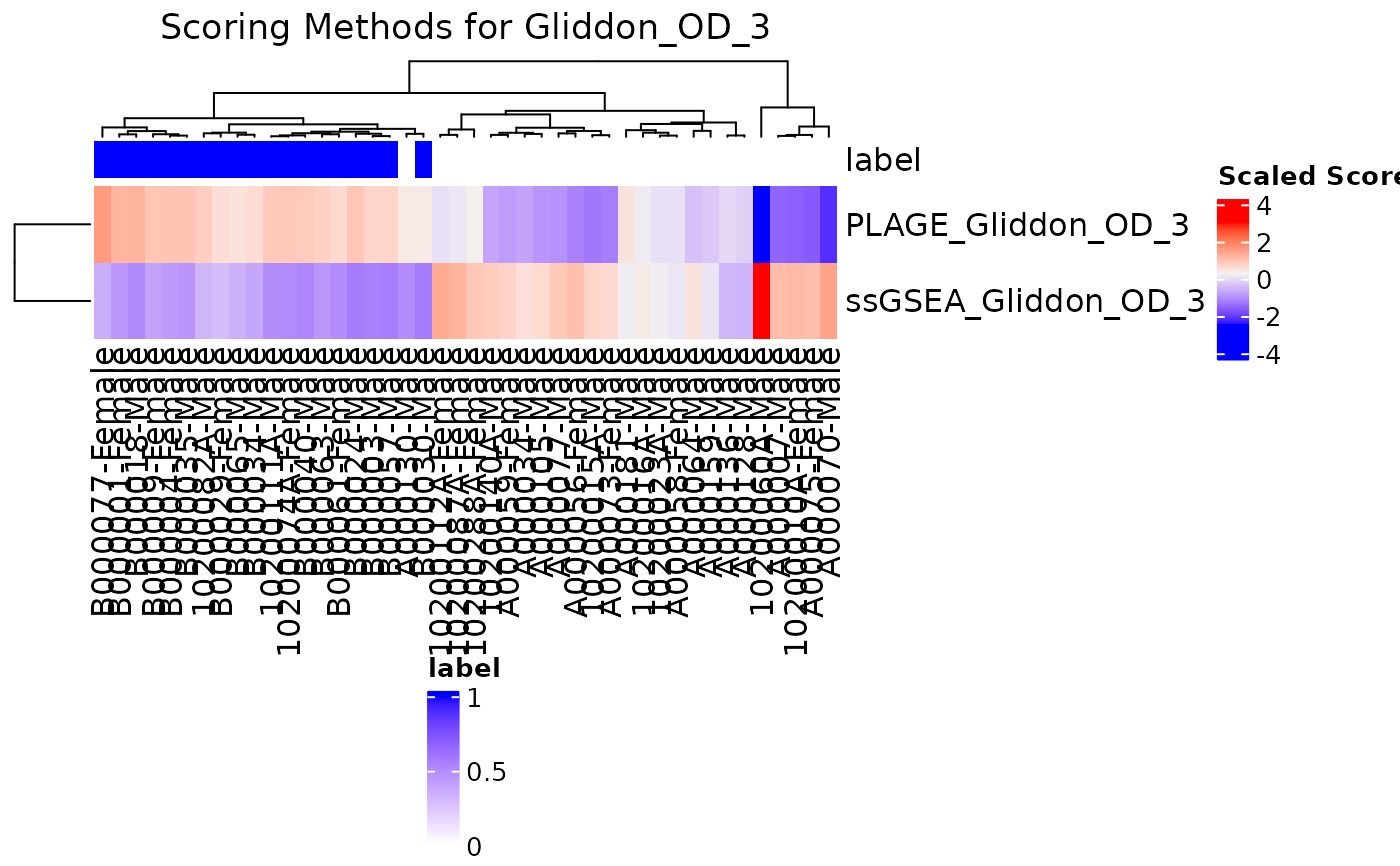
Compare scoring algorithms on a single signature via heatmap or boxplot.
Source:R/profile.R
compareAlgs.RdIt may be useful to compare the results of scoring across several different
scoring algorithms via a method of visualization, such as a heatmap. The
compareSigs function allows the input of a SummarizedExperiment
data object and conducts
profiling on each signature desired, and outputting a heatmap or boxplot
for each signature.
Usage
compareAlgs(
input,
signatures = NULL,
annotationColName,
useAssay = "counts",
algorithm = c("GSVA", "ssGSEA", "ASSIGN", "PLAGE", "Zscore", "singscore"),
showColumnNames = TRUE,
showRowNames = TRUE,
scale = FALSE,
colorSets = c("Set1", "Set2", "Set3", "Pastel1", "Pastel2", "Accent", "Dark2",
"Paired"),
choose_color = c("blue", "gray95", "red"),
colList = list(),
show.pb = FALSE,
parallel.sz = 0,
output = "heatmap",
num.boot = 100,
column_order = NULL
)Arguments
- input
an input data object of the class
"SummarizedExperiment". Required.- signatures
a
listof signatures to run with their associated genes. This list should be in the same format asTBsignatures, included in the TBSignatureProfiler package. Ifsignatures = NULL, the default set of signaturesTBsignatureslist is used. For details, run?TBsignatures. If <2 genes in a signature are present in the sample, that signature will not be evaluated and will not be present in the resulting SE object. The default isNULL.- annotationColName
a character string giving the column name in
colDatathat contains the annotation data. Required.- useAssay
a character string specifying the assay to use for signature profiling when
inputis aSummarizedExperiment. Required only for input data of the classSummarizedExperiment. If null, the assay used will be"counts". The default isNULL.- algorithm
a vector of algorithms to run, or character string if only one is desired. The default is
c("GSVA", "ssGSEA", "ASSIGN", "PLAGE", "Zscore", "singscore"). NOTE: ASSIGN takes a long time to run and is not recommended for efficient use.- showColumnNames
logical. Setting
showColumnNames = TRUEwill show the column names (i.e. sample names) on the heatmap. The default isTRUE.- showRowNames
logical. Setting
showColumnNames = TRUEwill show the row names (i.e. signature names) on the heatmap. The default isTRUE.- scale
logical. Setting
scale = TRUEscales the signature data. The default isFALSE.- colorSets
a vector of names listing the color sets in the order that they should be used in creating the heatmap. By default, this function will use the color sets in the order listed in
Usagefor annotation information. You may replace the default with the same collection of sets in order that you want to use them, or provide custom color sets with thecolListparameter.- choose_color
a vector of color names to be interpolated for the heatmap gradient, or a
colorRampfunction produced bycirclize::colorRamp2. The default isc("blue", "gray95", "red").- colList
a named
listof named vectors specifying custom color information to pass toComplexHeatmap::Heatmap(). The list should have as many elements as there are annotation columns, and each element name should correspond exactly with the name of each annotation column. The colors in the vector elements should be named according to the levels of the factor in that column's annotation data if the annotation is discrete, or it should be produced withcirclize::colorRamp2if the annotation is continuous. By default,ColorBrewercolor sets will be used. See the the parametercolorSetsfor additional details.- show.pb
logical, whether warnings and other output from the profiling should be suppressed (including progress bar output). Default is
FALSE.- parallel.sz
an integer identifying the number of processors to use when running the calculations in parallel for the GSVA and ssGSEA algorithms. If
parallel.sz = 0, all cores are used. The default is0.- output
a character string specifying whether the outputted plot should be a
"heatmap"or"boxplot". The default is"heatmap".- num.boot
an integer indicating the number of times to bootstrap the data.
- column_order
a vector of character strings indicating the order in which to manually arrange the heatmap columns. Default is
NULL, such that column order is automatically determined via clustering.
Examples
compareAlgs(TB_indian,
signatures = TBsignatures[c("Gliddon_OD_3")],
annotationColName = "label",
algorithm = c("ssGSEA", "PLAGE"),
scale = TRUE, parallel.sz = 1, output = "heatmap")
#> Parameter update_genes is TRUE. Gene names will be updated.
#> Running ssGSEA
#> ! 1 genes with constant values throughout the samples
#> Running PLAGE
#> ! 1 genes with constant values throughout the samples
#> ! Genes with constant values are discarded